Efficient Machine Learning Force Field for Large-Scale Molecular Simulations of Organic Systems
Junbao Hu, Liyang Zhou*, Jian Jiang*
arXiv:2312.09490
To address the computational challenges of ab initio molecular dynamics and the accuracy limitations of empirical force fields, the introduction of machine learning force fields has proven effective in various systems including metals and inorganic materials. However, in large-scale organic systems, the application of machine learning force fields is often hindered by impediments such as the complexity of long-range intermolecular interactions and molecular conformations, as well as the instability in long-time molecular simulations.
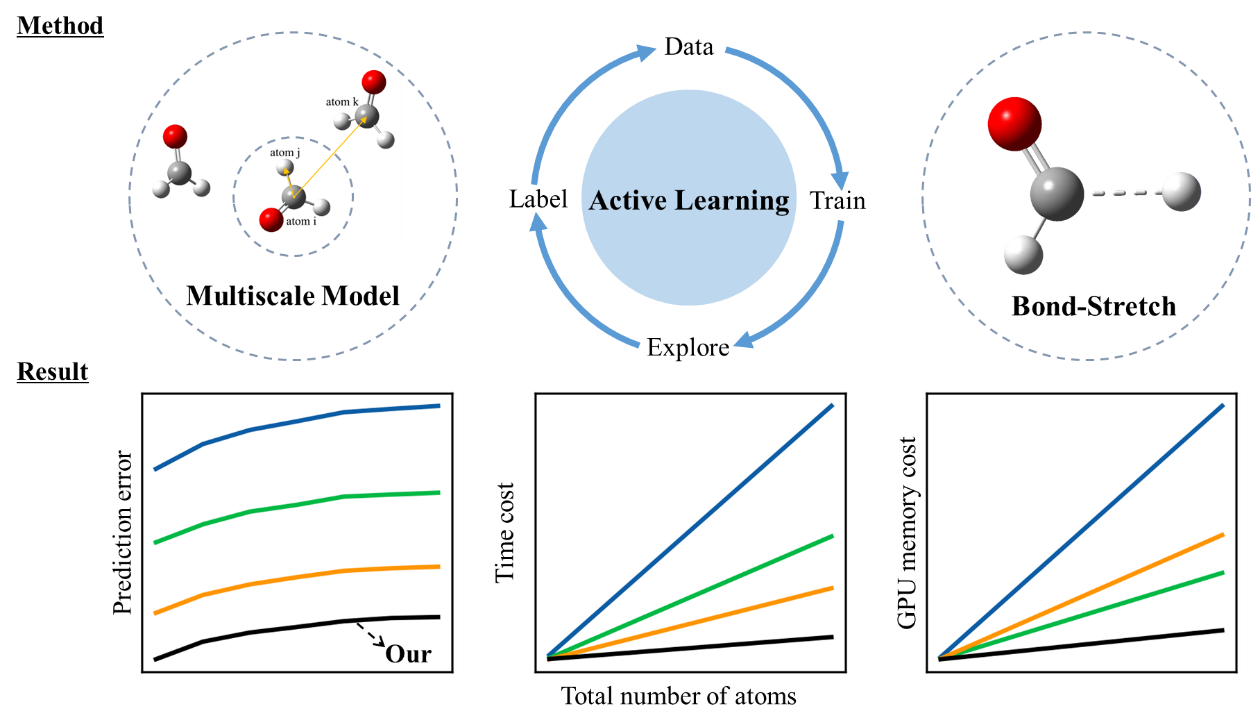
Therefore, we propose a universal multiscale higher order equivariant model combined with active learning techniques, which is efficient in capturing the complex long-range intermolecular interactions and molecular conformations. Compared to existing equivariant models, our model achieves the highest predictive accuracy, and magnitude-level improvements in computational speed and memory efficiency.
In addition, a bond length stretching method is designed to improve the stability of long-time molecular simulations. Utilizing only 901 samples from a dataset with 120 atoms, our model successfully extends high precision to systems with hundred thousand atoms. These achievements guarantee high predictive accuracy, fast simulation speed, minimal memory consumption, and robust simulation stability, satisfying the requirements for high-precision and long-time molecular simulations in large-scale organic systems.
文章链接:https://arxiv.org/abs/2312.09490